Overview of the prediction method for SMB gene clusters. (a

Download scientific diagram | Overview of the prediction method for SMB gene clusters. (a) Broken lines represent homologous gene pairs between two genomes. Each pair of 'x i1 '-'y j1 ', 'x i2 '-'y j2 ', 'x i4 '-'y j4 ', 'x i5 '-'y j8 ', 'x i6 '-'y j5 ' and 'x i8 '-'x j7 ' represents a homolog. The x i and y j represent genes in the first and the second genomes, respectively. (b) The genes were aligned in the genome using the Smith-Waterman algorithm (Param2 ¼ 21). Pairs of contiguous genes from 'x i1 ' to 'x i8 ' in genome 1 and from 'y j1 ' to 'y j7 ' represent an example identified as a seed for predicting a gene cluster (R 0 or other seed regions). (c) The seed was extended until the prescribed length (Param3 ¼ 35). The symbols l x and l y represent the numbers of genes added to the seed region of the first and the second genomes, respectively. X and Y represent extended clusters in the first and the second genomes, respectively. (d) The boundaries were corrected (Param4 ¼ 21), and a pair of candidate gene clusters, 'x i1 ' through 'x i8 ' and 'y j1 ' through 'y j8 ', was identified. The symbols i begin and i end represent the locations of the genes at the beginning and end, respectively, of the cluster in the first genome. The symbols j begin and j end represent the corresponding gene locations in the second genome. The CB value is the sum of the maximum scores for the upstream and the downstream boundaries of a predicted cluster. The integers are indicated as an example for the particular alignment of clusters represented in this figure. (e) Synteny analysis was performed to distinguish the SMB gene cluster from the syntenic block (SB). The SB, a subset of X and Y, represents a set of genes aligned to create a contiguous block of orthologous gene pairs located within the defined distance between neighboring genes (Param5 ¼ 10 kb). The above parameters are examples and not necessarily those used for the actual analyses. from publication: Motif-Independent Prediction of a Secondary Metabolism Gene Cluster Using Comparative Genomics: Application to Sequenced Genomes of Aspergillus and Ten Other Filamentous Fungal Species | Despite their biological importance, a significant number of genes for secondary metabolite biosynthesis (SMB) remain undetected due largely to the fact that they are highly diverse and are not expressed under a variety of cultivation conditions. Several software tools | Secondary Metabolism, Multigene Family and Comparative Genomics | ResearchGate, the professional network for scientists.
Functional annotation of genes in the two putative NRPS clusters. Two

Overview of the prediction method for SMB gene clusters. (a) Broken

A review of computational tools for generating metagenome-assembled genomes from metagenomic sequencing data. - Abstract - Europe PMC
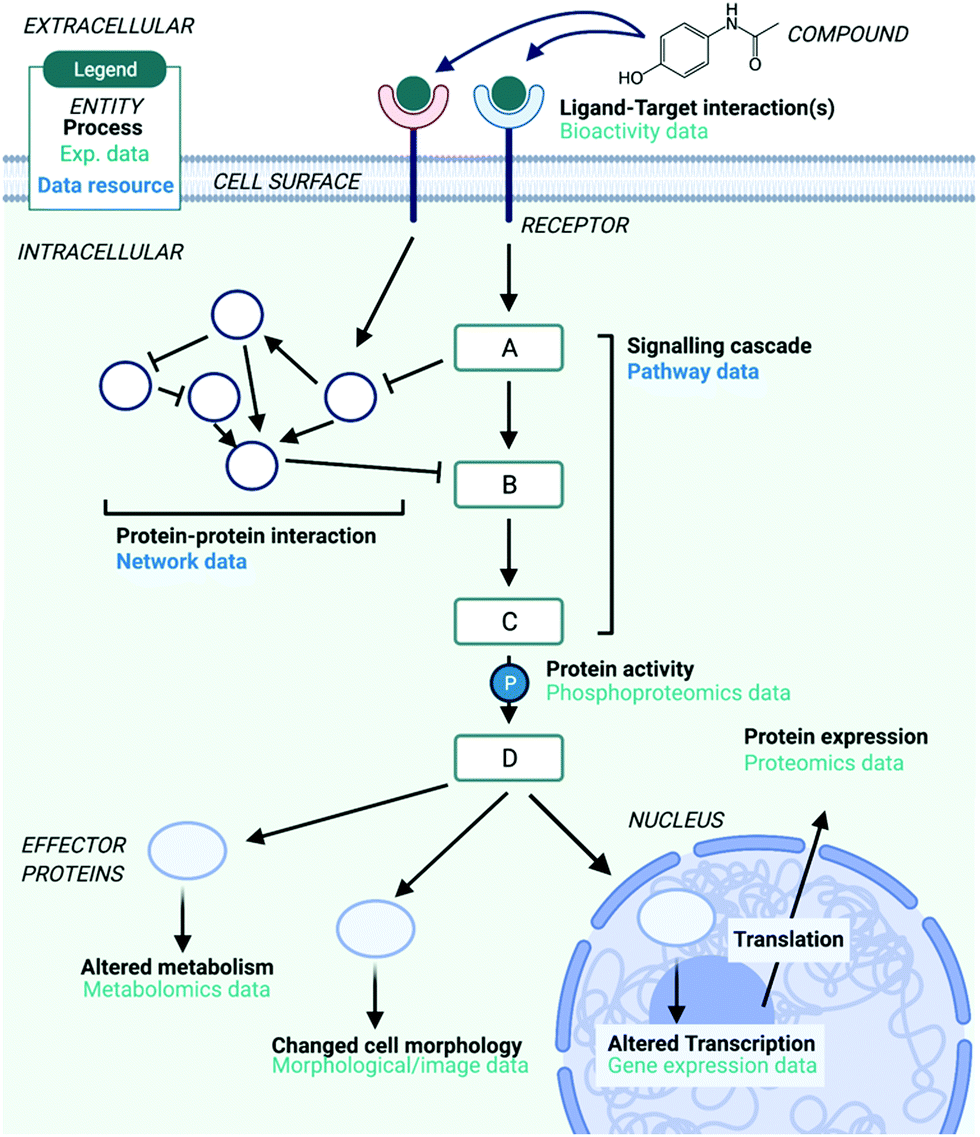
Computational analyses of mechanism of action (MoA): data, methods and integration - RSC Chemical Biology (RSC Publishing) DOI:10.1039/D1CB00069A

Figure 1 from antiSMASH 3.0—a comprehensive resource for the genome mining of biosynthetic gene clusters

Toward an integrated map of genetic interactions in cancer cells
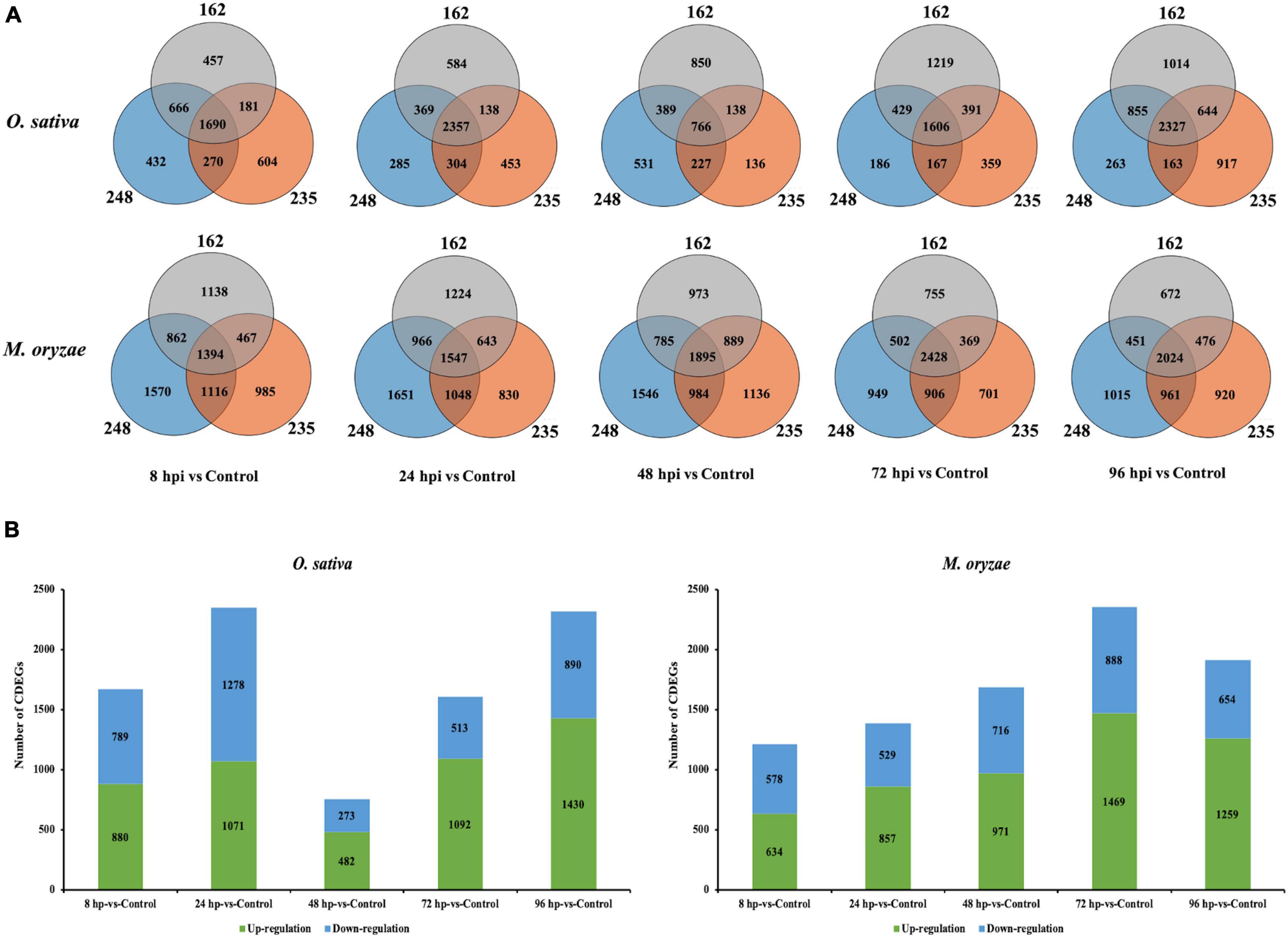
Frontiers Identification of Differentially Expressed Genes Reveal Conserved Mechanisms in the Rice-Magnaporthe oryzae Interaction

Workflow for the prediction of gene clusters in F. graminearum. Based

Detection methods for secondary metabolite biosynthetic gene clusters.
Evidence of horizontal gene transfer and regulation of predicted C47